联系我们
010-69739712

重组腺相关病毒(recombinant adeno-associated virus, rAAV) 载体与其他病毒载体相比, 具备感染能力强、可持续表达目的基因、无致病性和非基因组整合等优点, 已成为体内基因治疗的主要病毒载体。
本文总结了rAAV基因治疗产品的最新研究进展, 从原材料、生产工艺及质量控制等方面对此类产品的药学评价考虑要点展开讨论, 以期促进此类产品的临床转化与应用。同时对3个国外已上市产品的药学评价考虑要点及常见问题进行讨论。
rAAV基因治疗产业发展现状
据不完全统计, 目前至少有238 个rAAV基因治疗产品正在开展临床试验, 3 个rAAV基因治疗制品已在欧美注册上市(表1)。
表1 rAAV基因治疗产品的临床应用
导读:
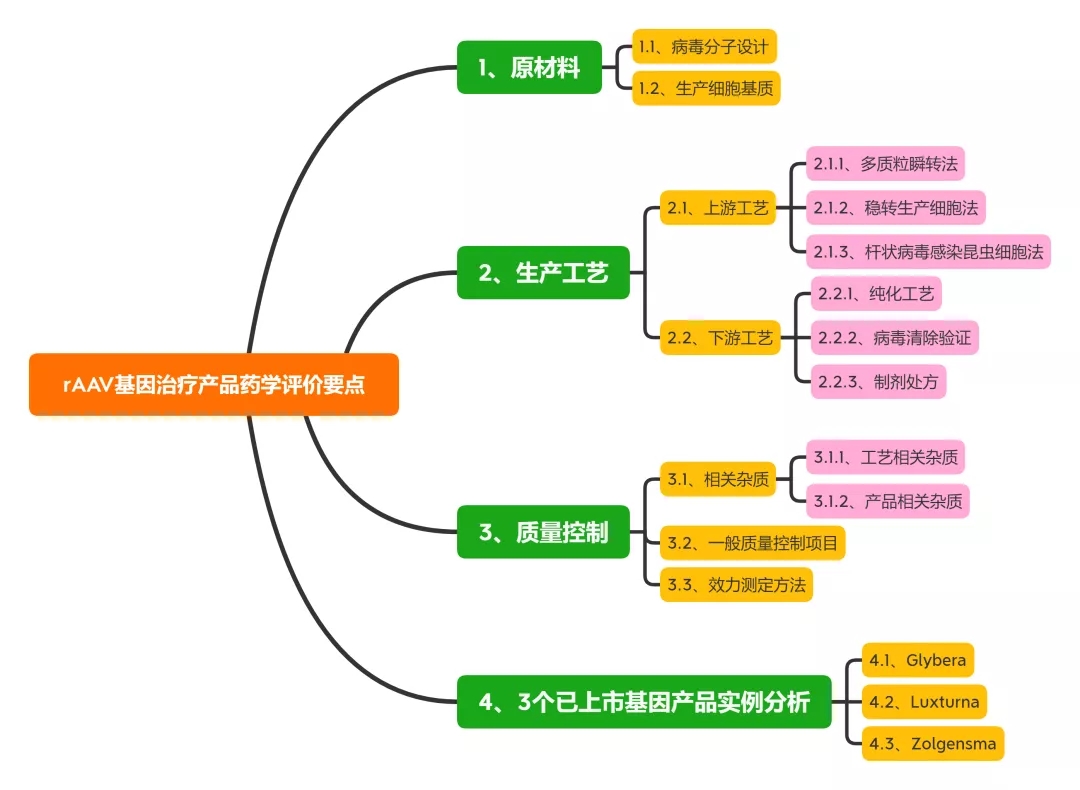
1、原材料相关研究内容与评价
1.1、病毒分子设计
rAAV的载体设计应基于临床有效性和安全性, 一般为靶向特定组织或细胞, 删除与毒力、致病性或复制能力相关的基因, 以确保制品的安全性。同时, 设计应考虑载体基因组的大小、包装效率和表达效率, 并尽可能减少与体内相关病毒的同源性, 以避免形成复制型病毒。构建病毒的质粒应考虑抗生素抗性基因引入的风险, 一般不得使用氨苄青霉素抗性基因。
1.2、生产细胞基质
目前产业界用于rAAV生产的细胞基质既有哺乳动物细胞(HEK293、BHK)、人肿瘤细胞系(HeLa、A549),也包括昆虫细胞系(SF9) 等。
包装细胞应进行充分的鉴定并建库。鉴定项目一般包括鉴别、纯度、基因型/表型、成瘤性/致瘤性、遗传稳定性和引入序列等。除常规无菌、真菌和支原体外, 尤其要关注细胞基质的种属特异性病毒。如: HEK293 细胞应检定CMV、HIV-1/2、HILV-1/2、HH-V6/8、JV virus、BK virus、EBV、parvovirusB19、HBV、HPV和HCV等病毒; SF9 细胞应检定螺原体、弹状病毒等昆虫病毒。
用人肿瘤细胞系作为包装细胞, 还应充分评估其致瘤性和成瘤性的风险, 以及致瘤病毒的感染情况等, 如: HeLa 细胞为来自人子宫颈癌组织的肿瘤细胞, 已知含有50 个拷贝数的HP
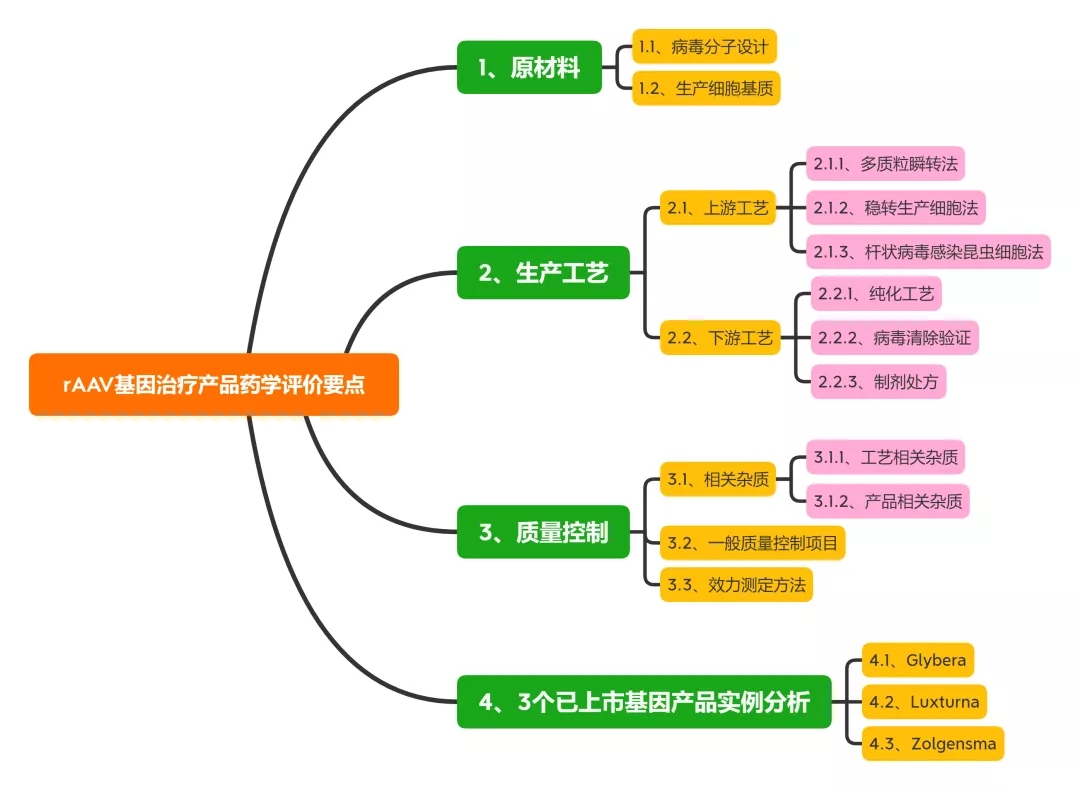
2、生产工艺相关研究与评价
2.1、AAV制品上游工艺
2.1.1、多质粒瞬转法
最早的rAAV 生产工艺采用质粒、辅助病毒感染包装细胞生产。目前采用较多的是三质粒共转染HEK 293 生产工艺, HEK293 细胞中含有腺病毒(adenovirus, AdV) 的E1a 和E1b 基因, 共转染转移质粒(含目的基因和ITR序列)、结构质粒(含rep/cap 基因) 和辅助质粒( 复制病毒基因E2A, E4,VARNA 等), 48~72 h 后即可重组包装rAAV。
该方法适用于多种血清型rAAV, 发酵产率一般可达每毫升1014 V.G. (vector genome), 一般可满足早期临床试验rAAV用量(<1015 V.G.)。但是, 受贴壁培养方式所限, 多质粒瞬转工艺较难满足商业化生产需求(>1016 V.G.)。
2.1.2、稳转生产细胞法
构建含有辅助基因rep/cap和目的基因的稳转HeLa 细胞系, 经腺病毒感染后也可包装出rAAV 病毒。尤其是采用可悬浮培养的HeLa S3细胞系, 稳转细胞生产法可直接放大至2 000 L规模。与之类似, 构建含有ICP27 基因的BHK 细胞,经转染含有辅助基因和目的基因的复制缺陷型d27.1HSV后, 也可高效表达rAAV。
但是, 稳转细胞法的主要缺点在于, 细胞构建和遗传稳定性研究较为耗时,且生产过程中使用辅助病毒存在病毒安全性风险。
2.1.3、杆状病毒感染昆虫细胞法
杆状病毒具有高度的种属特异性, 不感染脊椎动物, 能将rAAV基因和辅助功能的反式作用元件转移至SF9 昆虫细胞中。因此, 近年也开始应用于rAAV 的大规模生产。该方法的优势在于杆状病毒生物安全性好, 感染效率高, 生产工艺便于放大。但质量控制项目中也应考虑杆状病毒及DNA相关物质残留。
药学评价要点:
对于rAAV 上游生产工艺, 药学评价建议关注关键工艺的工艺开发与验证, 如采用多质粒瞬时转染或加入辅助病毒等工序中, 应通过工艺开发与验证说明关键工艺参数的控制范围及中间体验收标准, 如不同载体配比、转染试剂用量、辅助病毒与生产细胞的感染复数等。
2.2、AAV制品下游工艺
2.2.1、纯化工艺
rAAV 纯化工艺一般包括细胞培养液收获、化学法/物理法裂解细胞、benzonase 酶消化去除核酸物质、多步层析和密度梯度离心、置换制剂处方缓冲液等工序, 其中, 亲和层析模拟细胞受体的结合作用捕获rAAV。如: 硫酸肝素填料可特异性结合rAAV2, AVB琼脂糖填料可结合1、2、3、5 和8 等多种血清型rAVV[22]。对于空载体的去除, 目前多采用碘克沙醇和氯化铯超速离心法。
值得关注的是, 中国药典相关总论中明确指出,“除另有证明其合理性外, 不得使用氯化铯-溴化乙锭密度离心法进行基因治疗产品的纯化”。
2.2.2、病毒清除验证
如上所述, 采用杆状病毒-昆虫细胞或稳转细胞系(HeLa) 生产工艺时会使用杆状病毒或辅助病毒(HSV、Ad5 等)。因此, 下游工艺应增加特定病毒去除/灭活工序。
如: 采用表面活性剂TritonX-100 (0.5%, v/v) 和增加病毒过滤工序去除收获液中残留杆状病毒; 采用加入表面活性剂、低pH值灭活等工序去除HSV病毒; 采用离子交换法、短时间加热(52 ℃、10 min) 和纳滤等工序去除腺病毒等。
2.2.3、制剂处方
由于rAAV衣壳蛋白对温度和pH值变化并不敏感, rAAV 制品的制剂处方开发相对简单。通常惯用在盐溶液中加入200 mmol·L-1硫酸镁或0.001%泊洛沙姆F68 避免产生聚体, 基本可支持长期保存(-65 ℃) 和运输稳定性。
评价要点:
对于rAAV 下游生产工艺, 药学评价建议结合产品相关杂质、过程相关杂质的去除效率评估纯化工艺合理性与稳健性。对于使用辅助病毒的生产工艺应结合收获液中病毒含量, 进行病毒去除/灭活工艺验证,综合评估病毒的残留安全性。
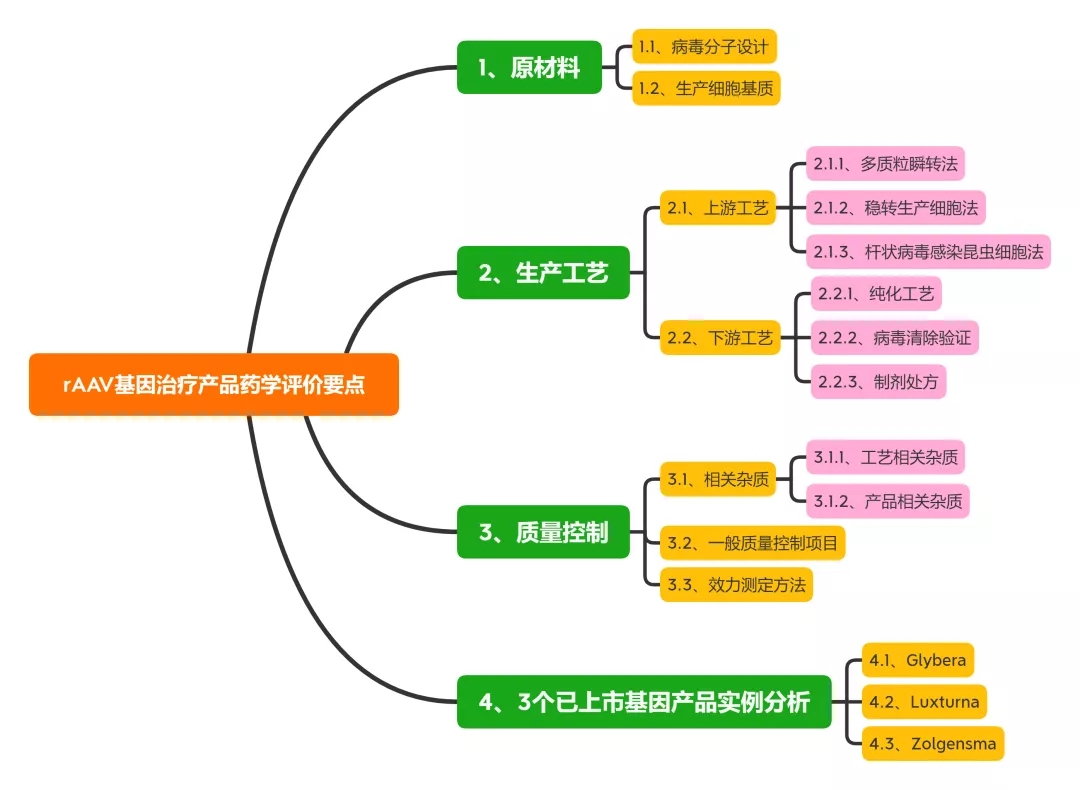
3、病毒清除验证
3.1、相关杂质
3.1.1、工艺相关杂质
rAAV 制品的工艺相关杂质包括病毒包装与纯化工艺中引入的宿主蛋白、宿主DNA、辅助病毒、质粒、血清和氯化铯等组分。由于病毒包装细胞通常含有致癌基因, 如HEK293 细胞内含有E1A 的腺病毒基因, HeLa 细胞内的E6、E7 致癌基因。因此, 一般对于宿主细胞残留DNA 含量应小于10 ng/Dose, 且残留DNA片段应小于200 bp。
3.1.2、产品相关杂质
rAAV 制品的相关杂质包括未包装基因的空病毒、包装错误基因(宿主DNA、不完整目的基因、辅助病毒基因等) 的病毒、无感染活性的病毒颗粒、聚体或氧化形式的病毒颗粒等。
这些产品相关杂质不仅不能实现目的基因在靶细胞的表达, 还可在临床上引起免疫原性或基因毒性。如: 多质粒顺转体系中, 一般空病毒占比可以达到50%~98%。空病毒不具感染能力, 且容易形成病毒聚体和降解, 引起体内免疫反应。因此, 质量研究中应采用A260/A280、透射电镜、分析性离心和质谱等技术检测空病毒含量。
复制型腺相关病毒(replication-competent AAV,rcAAV) 是由于同源/非同源重组发生后, rAAV病毒同时包装rep 和cap/AAP等基因所产生的产品相关杂质。
rcAAV在辅助病毒存在的条件下可以进行复制扩增。rcAAV的检测一般采用在辅助病毒存在下使用敏感细胞进行扩增, 细胞裂解液经过多次扩增传代后, 采用Southern blot 和qPCR法测定rep 或cap 基因。如: 已进入临床试验的rAAV 制品scAAV2/8-LP1-hFIXco 采用qPCR 法控制rcAAV 含量限度低于1/2.25×106 。目前也有报道称采用优化后的多质粒瞬转工艺,rcAAV含量限度可低于1/108 V.G.。
3.2、一般质量控制项目
rAAV病毒生产质量控制包括过程控制与终产品放行检测(表2)。
如: 病毒包装用质粒应进行鉴别、含量、纯度、宿主细胞DNA残留、转染效率、细菌内毒素、无菌等质量控制;
病毒收获液应控制外源因子(无菌、支原体等)、外源病毒、目的病毒等检测;
rAAV原液与制剂放行项目一般包括外观、理化性质(pH值、渗透压)、病毒滴度(物理滴度、感染滴度)、纯度(蛋白纯度、吸光度比值、宿主DNA残留、质粒DNA残留、核酸酶残留)、效力(目的基因表达、体外活性、体内活性)、安全性(内毒素、无菌、rcAAV)。
此外, 对于眼部用药的rAAV 制剂, 内毒素含量应不高于2.0 EU/dose/eye 或0.5 EU/mL, 应按照眼用制剂控制不溶性微粒(USP<789>)、产品的放行检测应包括配置后产品等。

3.3、效力测定方法
对于rAAV 制品的效力测定方法, 应体现其病毒物理滴度、感染活性及生物活性。
一般在临床试验早期可采用qPCR 法测定病毒基因组, 敏感细胞或靶向细胞测定其感染能力和目标产物表达能力表征产品效力。
关键性临床试验开展后应进一步开发反映产品作用机制的体外酶活法或体内功能实验法。如:SPK-RP65 在进入Ⅲ期临床后, 通过转染指示细胞(HEK293-LRAT) 后测定RPE65 蛋白酶促反应产物视黄醇含量来计算产品效力。
值得指出的是, 对于qPCR法测定病毒基因组应采用线性DNA绘制标准曲线, 若采用超螺旋DNA作为参照品, 载体基因组含量测定值显著高于真实值。
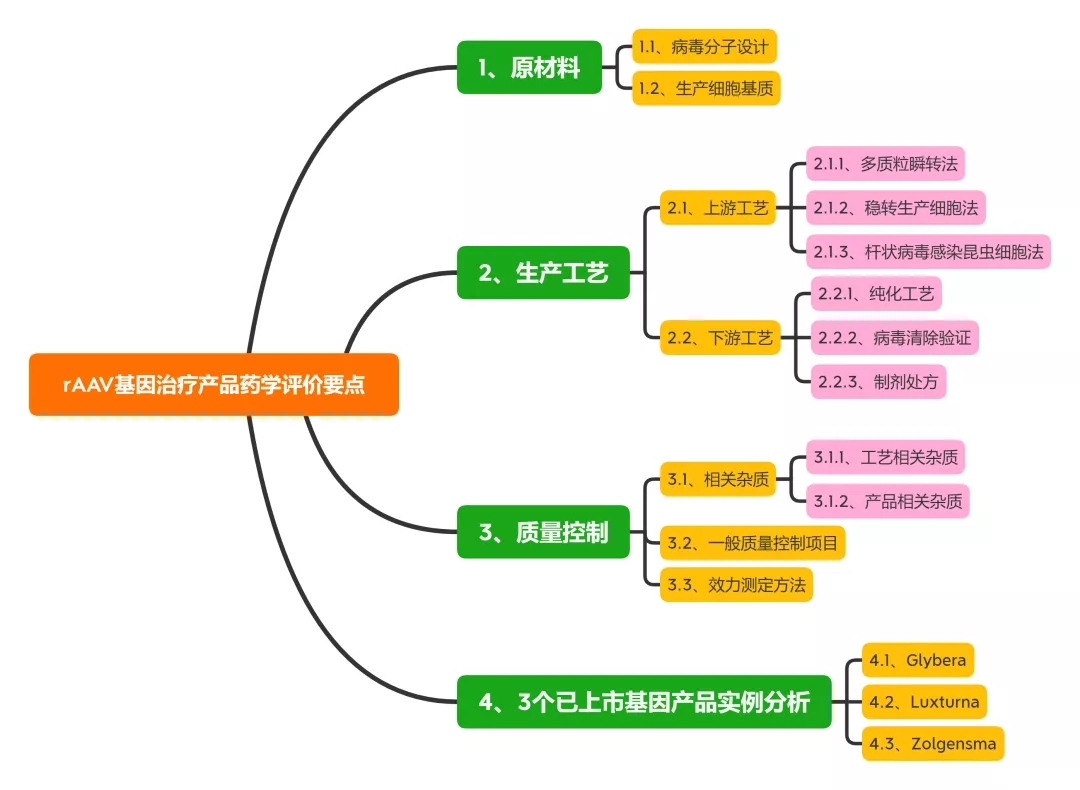
4、3个已上市基因产品实例分析
目前, 国际上仅有3个rAAV基因治疗产品(glybera、luxturna 和zolgensma) 获批上市, 国内此类产品多处于研发早期阶段, 工业界与监管方对于rAAV 基因治疗产品的药学研究与评价均缺乏实践经验。下文拟结合国外已上市产品审评报告中披露信息, 对此类产品的药学评价考虑要点及常见问题进行讨论。
4.1、Glybera
Glybera (alipogene tiparvovec, AAV1-LPLS447X)是Amsterdam Molecular Therapeutics 公司开发的携带人LPLS447X 基因的rAAV2 型基因治疗产品, 临床上肌肉注射用于治疗成人脂蛋白脂肪酶缺陷症。
本品工艺开发过程中存在重大工艺变更: 第一代生产工艺(AMT-010) 采用HEK293 表达系统, 商业化生产工艺(ATM-011) 则采用杆状病毒-昆虫细胞系统表达, 因此开展了药学和非临床可比性研究。
下游工艺中采用工艺规模缩小模型对包膜病毒、非包膜病毒去除能力进行了工艺验证。
质量研究对 3 批连续生产批次制品进行了充分的表征研究, 其中包括: 组分(基因组完整性/大小、蛋白质分析、分子质量、衣壳蛋白); 物理性质(颗粒大小、病毒颗粒糖基化修饰); 一级结构(序列确证、蛋白鉴定); 高级结构(透镜电镜、分析超速离心); 生物活性(感染性颗粒、比活、效力) 等。
EMEA审评认为, 由于细胞收获液中含有大量感染性杆状病毒, 且下游工艺不能提供足够的病毒去除效果, 要求申请人补充提供临床注射杆状病毒残留DNA的风险分析报告, 并建议产品放行检测项目中增加感染性杆状病毒残留和rcAVV等检查项目。
并且, EMEA审评建议产品上市后生产工艺应进一步增加杆状病毒去除工序(如病毒过滤), 后期应提高杂质(包装细胞DNA、残留Rep/Cap 基因、rcAAV、感染性杆状病毒等残留) 检测方法灵敏度。
4.2、Luxturna
Luxturna (voretigene neparvovec-rzyl, SPK-RPE65)系美国Spark Therapeutics 公司研发的携带人RPE65基因rAAV-2 型基因治疗制品, 临床上视网膜下注射用于治疗先天性黑朦症。
产品采用三质粒共转染贴壁HEK293 细胞, 滚瓶生产工艺。生产工序包括: 细胞扩增、转染、培养基置换、收获培养液、切向流过滤、均质、离子交换色谱、密度梯度离心、制剂缓冲液置换和过滤等;
本品Ⅲ期临床试验前曾发生场地、包装容器等工艺变更, 采用“Side-by-side”法对产品质量的方法进行产品可比性研究。
产品放行检测项目包括: 理化(外观、pH值、浓度、可抽取体积)、鉴定(目的基因)、含量(基因组浓度)、效力(目的基因表达、体外活性)、纯度和安全性项目(内毒素、颗粒物、无菌) 等, 质量研究中对宿主DNA、质粒DNA、E1A 基因和牛血清蛋白等工艺相关杂质残留进行控制。
FDA审评认为, 本品上市后应继续完成包装细胞HKE293 稳定性及产品实时稳定性等研究。
4.3、Zolgensma
Zolgensma (onasemnogene abeparvovec, AVXS-101)系AveXis 公司开发的携带运动神经元蛋白1 (survival motor neuron, SMN) 基因的rAAV9 型基因治疗产品,临床上静脉注射治疗儿童脊髓肌肉萎缩症。
本品在工艺开发过程中, 结合目标质量属性采用风险评估方法确定本品关键质量属性, 采用工艺规模缩小模型确定工艺设计空间并进行工艺验证;
本品在关键性临床试验开展前、注册上市前, 先后发生场地、工艺和制剂处方等变更。
FDA 审评判定工艺变更前后产品纯度和VG 单位效力保持一致; 本品的“效力”检测项中既包括定量检测靶细胞SMN蛋白表达水平, 也包括体内半定量法检测小鼠“生存率”。
值得关注的是, 由于本品在І 期临床试验开展过程中“含量”分析方法缺乏精确度与准确度, 44 个月后采用更新的方法重新修正临床给药剂量。此外, 在本品稳定性实验中, 观测到“含量”和“效力”有所下降。
